Class III medical devices represent the highest risk classification under Regulation (EU) 2017/745 (MDR). These are devices that are life-supporting, life-sustaining, implanted for long-term use, or present a high degree of invasiveness or complexity. Because they pose the most serious potential risks to patient health and safety, they are subject to the most rigorous regulatory scrutiny and control.
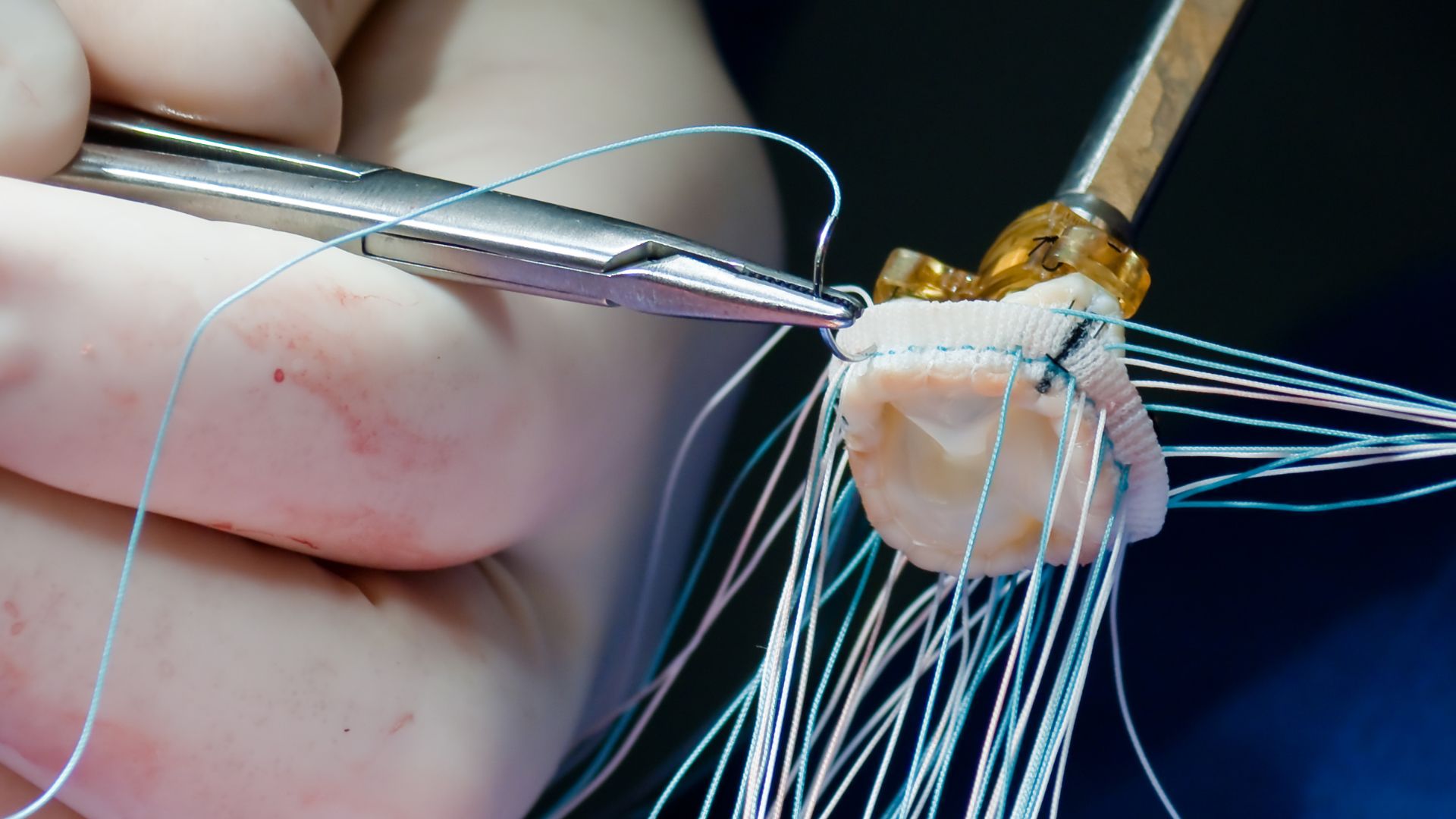
Examples of Class III devices include implantable pacemakers, prosthetic heart valves, spinal disc replacements, drug-eluting stents, and deep-brain stimulators. These devices often involve advanced materials, complex engineering, or integration with medicinal substances and biological components. As a result, they require a high degree of regulatory maturity from manufacturers, including robust design and risk controls, comprehensive clinical validation, and tight postmarket surveillance.
This article explores the full spectrum of EU requirements for Class III devices—from initial classification and clinical strategy to conformity assessment, technical documentation, economic operator obligations, and lifecycle management. It is intended for regulatory professionals, manufacturers, and quality leaders preparing to bring Class III devices to the EU market or maintain compliance post-certification.
What Makes a Device Class III Under MDR?
Under MDR Annex VIII, the classification of a device as Class III is based primarily on its invasiveness, duration of contact, interaction with critical physiological systems, and incorporation of substances that present additional risks. Rules such as Rule 8 (implantable devices), Rule 13 (medicinal substance devices), Rule 14 (devices for contraception or prevention of sexually transmitted diseases), and Rule 21 (substance-based devices) frequently capture Class III products.
For instance, an implantable prosthetic valve remains in the body long-term, directly supports a life-sustaining function, and is in constant interaction with blood. These characteristics escalate the risk profile, thereby requiring a Class III designation. Similarly, devices that administer systemic pharmaceutical agents or involve human- or animal-derived substances automatically trigger Class III classification.
Correct classification is essential not only for selecting the appropriate regulatory pathway but also for risk communication, CE marking validity, and postmarket vigilance obligations.
Conformity Assessment Procedures: Annex IX and Beyond
Conformity assessment for Class III devices requires deep Notified Body involvement and strict documentation and clinical scrutiny. The most common assessment route is via Annex IX, which involves a full QMS audit, technical documentation review, and an in-depth evaluation of the design dossier.
The process includes an initial QMS certification followed by a design examination specific to each Class III product. Unlike lower-class devices where sampling may occur, all Class III device designs are examined individually. This ensures that the manufacturer can demonstrate conformity with all applicable General Safety and Performance Requirements (GSPRs) for that specific product.
Manufacturers can also pursue a combined Annex IX and Annex X route or use Annex XI (Part A and B) for product verification, but these are typically reserved for unique manufacturing or product configurations. Regardless of the path chosen, Notified Bodies perform annual surveillance audits, unannounced inspections, and monitor clinical and PMS data to confirm continued compliance.
Clinical Evaluation and Investigation Requirements
Annex XIV of the MDR mandates comprehensive clinical evaluation for Class III devices. This must be based on high-quality data, including clinical investigations conducted in accordance with ISO 14155 and MDR Chapter VI. Manufacturers cannot rely solely on equivalence unless they have full access to technical documentation and demonstrate identical device characteristics, biological effects, and clinical use.
Clinical investigations are often required and must be designed with statistical justification, defined endpoints, risk-benefit metrics, and follow-up timelines. Data from these studies are used to build the Clinical Evaluation Report (CER), which forms a key part of the technical documentation.
Postmarket clinical follow-up (PMCF) is also mandatory and must be defined in a PMCF Plan that aligns with the Clinical Evaluation Plan. PMCF may involve observational studies, registry participation, or long-term outcome tracking, especially for implantable or novel devices. The PMCF Evaluation Report must be updated regularly and submitted alongside the Periodic Safety Update Report (PSUR).
Technical Documentation and SSCP for Class III
The technical documentation for Class III devices is expansive and must comply with Annex II and III of the MDR. It includes device description and specifications, manufacturing processes, quality and risk management files, performance and safety testing, software validation, and biological compatibility data.
A Class III device must also have a Summary of Safety and Clinical Performance (SSCP), which is reviewed and approved by the Notified Body. The SSCP is made publicly available via EUDAMED and must be written in layperson terms for patients and clinicians.
The SSCP summarizes the device’s intended purpose, indications, contraindications, target populations, clinical outcomes, residual risks, and handling instructions. It is designed to improve transparency and user understanding and is subject to ongoing updates.
Quality Management System and Design Controls
A mature and fully integrated QMS is a prerequisite for Class III device manufacturers. This includes design control documentation, risk management per ISO 14971, and alignment with ISO 13485:2016. The QMS must ensure traceability across the product lifecycle—from initial requirements and design inputs through production, batch release, PMS, and eventual retirement.
Design controls must include usability engineering (IEC 62366), software lifecycle compliance (IEC 62304), and, where applicable, cybersecurity risk controls. Manufacturers must implement formal design reviews, design verification and validation (V&V), and maintain a design history file (DHF).
In Class III, any design change—even minor—may require notification to the Notified Body or a re-review of the design dossier. Change control, configuration management, and internal communication systems must be capable of managing this rigor and traceability.
Postmarket Surveillance, Vigilance, and PSUR
The postmarket obligations for Class III devices are among the most intensive. Manufacturers must maintain a PMS system that collects, analyzes, and reports real-world data about device performance, user complaints, adverse events, and clinical outcomes.
The Periodic Safety Update Report (PSUR) must be updated and submitted annually to the Notified Body. It must summarize PMS results, PMCF findings, risk-benefit updates, and cumulative sales and usage data. Any emerging safety signals must be accompanied by CAPA plans or design modifications.
Vigilance reporting includes mandatory timelines for serious incidents and field safety corrective actions. FSCA reports must be followed by Field Safety Notices (FSNs) to affected stakeholders and coordinated through EU Competent Authorities. Failure to follow these processes can lead to suspension or withdrawal of the CE certificate.
Economic Operator and EUDAMED Responsibilities
Class III device manufacturers must engage a European Authorized Representative if they are not based in the EU. The EC REP must be listed on product labeling and registered in EUDAMED. Importers and distributors must verify that the device is CE-marked, labeled correctly, and accompanied by the correct documentation.
The Basic UDI-DI and UDI-DI must be registered in EUDAMED and appear on product packaging and SSCP documentation. Economic operators must be capable of tracing the product through distribution and responding to field actions. Supply chain traceability is a regulatory expectation—not a logistical option.
Strategic Planning and Market Access Readiness
Entering the EU with a Class III device demands strategic planning from product development through postmarket support. Manufacturers should conduct early gap assessments, pre-submission consultations with Notified Bodies, and feasibility studies to determine data sufficiency. Collaboration with clinical investigators, contract research organizations (CROs), and local regulatory advisors is essential.
Commercial teams must be trained in compliance-aligned messaging, and all claims in promotional materials must reflect the device’s SSCP and CER. Global harmonization strategies—such as leveraging U.S. FDA IDE or PMA data—must be carefully aligned with MDR expectations, recognizing that equivalence and reliance may be limited.
Class III as the Apex of EU Device Compliance
Navigating Class III medical device requirements under the MDR is complex and resource-intensive—but also essential for companies seeking to lead in high-impact healthcare technologies. Mastery of design, documentation, clinical validation, and postmarket responsiveness defines regulatory excellence at this level.
The path to CE certification for Class III devices is demanding, but it is also an opportunity to demonstrate the highest level of product assurance, organizational maturity, and patient commitment. For manufacturers that invest early and strategically in compliance, Class III readiness becomes more than a barrier—it becomes a competitive advantage and a statement of industry leadership.
Medical Devices
Latest Articles






Author
