
Bringing a pharmaceutical product to the European market requires far more than preparing a regulatory submission. Companies must understand the network of agencies and organizations that influence product approvals, quality standards, lifecycle management, and compliance obligations throughout Europe.
While the European Medicines Agency (EMA) remains central to many pharmaceutical approvals, Europe’s regulatory framework extends well beyond a single authority. Organizations such as the Heads of Medicines Agencies (HMA), the European Directorate for the Quality of Medicines & HealthCare (EDQM), Swissmedic, and the Medicines and Healthcare products Regulatory Agency (MHRA) each play important roles in shaping how medicines are reviewed, monitored, and maintained throughout their lifecycle. Together, these organizations illustrate how European regulation has evolved from independent national systems into a coordinated ecosystem balancing regulatory oversight, quality standards, and market access.
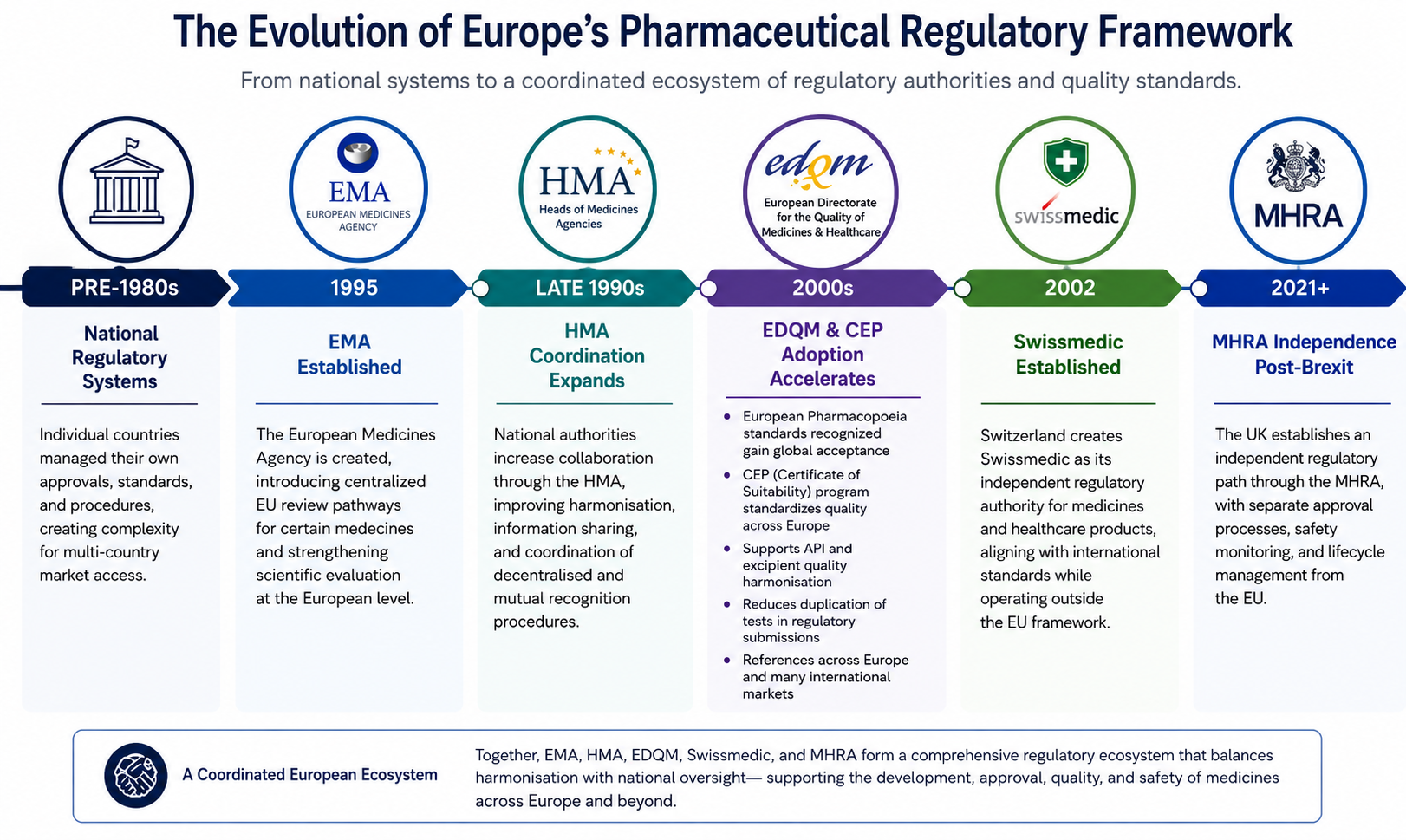
Together, these organizations illustrate how European pharmaceutical regulation has evolved from a collection of independent national systems into a highly coordinated regulatory ecosystem balancing harmonization with country-specific oversight.
The Evolution of Europe’s Regulatory Framework
Prior to the 1990s, pharmaceutical approvals in Europe were primarily managed by individual national authorities. Each country maintained its own regulatory procedures, review standards, and approval timelines, creating significant complexity for companies seeking access to multiple European markets.
As pharmaceutical development became increasingly global, regulators recognized the need for greater cooperation, stronger quality standards, and more efficient review processes. This led to the creation and expansion of several organizations designed to support coordination, harmonization, and regulatory consistency across the region.
The establishment of the EMA introduced centralized review pathways for certain medicines. At the same time, collaborative structures such as the HMA strengthened cooperation among national authorities, while EDQM expanded the role of harmonized pharmaceutical quality standards through the European Pharmacopoeia and Certificate of Suitability (CEP) system.
More recently, regulatory developments such as Brexit have further reshaped the landscape by creating an independent role for the MHRA outside the European Union framework.
Today, pharmaceutical companies must navigate a regulatory environment that includes multiple agencies and organizations, each with distinct responsibilities and regulatory influence.
The Role of the Heads of Medicines Agencies (HMA)
The Heads of Medicines Agencies (HMA) is a network composed of the national competent authorities responsible for regulating medicines across the European Economic Area (EEA).
Unlike the EMA, the HMA is not a centralized approval authority. Instead, it serves as a collaborative framework that helps coordinate regulatory activities between European national agencies.
The HMA was formally established during the 1990s as European regulatory cooperation expanded alongside the introduction of decentralized and mutual recognition procedures. As more pharmaceutical applications began involving multiple countries simultaneously, regulators recognized the need for stronger coordination between national authorities.
Over time, the HMA has become an important part of Europe’s regulatory structure by helping improve:
- Regulatory harmonization
- Information sharing between authorities
- Coordination during decentralized procedures
- Pharmacovigilance collaboration
- Regulatory data standardization
The organization also supports working groups and initiatives focused on improving consistency across European regulatory activities.
For pharmaceutical companies, the HMA plays an important behind-the-scenes role in helping national agencies align on scientific assessments, procedural standards, and regulatory expectations across Europe.
This coordination becomes especially important during:
- Decentralized Procedures (DCP)
- Mutual Recognition Procedures (MRP)
- Multi-country lifecycle management activities
- Cross-border pharmacovigilance coordination
Understanding the HMA framework can help companies better anticipate how national authorities collaborate throughout European review processes.
EDQM and CEPs: The Foundation of Pharmaceutical Quality Standards
While agencies such as the EMA, HMA, Swissmedic, and MHRA focus on regulatory oversight and market authorization activities, the European Directorate for the Quality of Medicines & HealthCare (EDQM) serves a different but equally important function.
Operating under the Council of Europe, EDQM is an independent European institution responsible for developing and maintaining the European Pharmacopoeia, one of the world’s most widely recognized collections of pharmaceutical quality standards.
One of EDQM’s most influential contributions to pharmaceutical regulation is the Certificate of Suitability (CEP) program.
A CEP demonstrates that the quality of a pharmaceutical substance can be adequately controlled according to the requirements of the relevant European Pharmacopoeia monograph. Rather than repeatedly submitting extensive substance data to multiple regulatory authorities, manufacturers can reference a CEP within their regulatory submissions.
Over time, CEPs have become a cornerstone of European pharmaceutical regulation and are frequently utilized by:
- Active pharmaceutical ingredient (API) manufacturers
• Excipient manufacturers (where applicable)
• Marketing authorization holders
• Regulatory authorities throughout Europe
• Companies pursuing international regulatory submissions
The increasing use of CEPs reflects an important shift in European regulation toward greater harmonization of pharmaceutical quality standards. For companies managing complex supply chains and multi-market submissions, CEPs can help improve consistency, reduce duplication, and support more efficient regulatory review processes.
Swissmedic – Switzerland’s Independent Regulatory Approach
Although Switzerland is not a member of the European Union, Swissmedic remains one of Europe’s most respected pharmaceutical regulatory authorities.
Swissmedic was established in 2002 following the consolidation of Switzerland’s regional regulatory oversight systems into a centralized national authority. Since then, the agency has continued expanding its international cooperation efforts while maintaining an independent regulatory framework outside the EU system.
Today, Swissmedic is responsible for:
- Reviewing and authorizing medicinal products in Switzerland
- Monitoring product safety
- Conducting inspections
- Overseeing manufacturing compliance
- Managing pharmacovigilance activities
Switzerland remains an important pharmaceutical market with a strong global industry presence, making Swissmedic a key authority for companies pursuing broader European commercialization strategies.
Although Swissmedic collaborates with international regulators and may participate in reliance or recognition pathways, its review procedures remain independent from the EMA.
For pharmaceutical companies, this means Swiss submissions often require careful planning around:
- Country-specific administrative requirements
- Local representation obligations
- Product labeling expectations
- Swiss-specific submission timelines
- Language and documentation requirements
Companies should avoid assuming that EU approval automatically guarantees alignment with Swiss regulatory expectations. Maintaining organized submission strategies across both EU and Swiss markets remains essential for efficient market access.
The MHRA – the United Kingdom’s Regulatory Transition
The Medicines and Healthcare products Regulatory Agency (MHRA) serves as the United Kingdom’s national regulatory authority for medicines, medical devices, and healthcare products.
The MHRA has long played a major role in European pharmaceutical regulation. However, its responsibilities evolved significantly following the United Kingdom’s departure from the European Union.
Prior to Brexit, many UK pharmaceutical approvals operated within the broader EMA framework. Since becoming an independent regulatory authority, the MHRA has introduced separate approval pathways, independent lifecycle management procedures, and new international reliance models designed to support regulatory flexibility within the UK market.
Today, the MHRA oversees:
- Marketing authorizations in the United Kingdom
- Drug safety and pharmacovigilance
- GMP inspections
- Clinical trial oversight
- Regulatory compliance monitoring
For pharmaceutical companies, Brexit created an entirely new regulatory dynamic between the EU and UK markets.
Organizations now managing both EU and UK commercialization strategies must maintain:
- Separate regulatory submissions
- Independent lifecycle management activities
- Distinct labeling compliance
- UK-specific regulatory timelines
- Parallel pharmacovigilance coordination
At the same time, the MHRA continues developing international collaboration and reliance pathways intended to accelerate access to medicines while maintaining regulatory oversight.
This evolving environment has made strong regulatory coordination increasingly important for companies operating across both jurisdictions.
Why Understanding These Authorities Matters
Although the HMA, Swissmedic, and MHRA operate differently, each plays an important role in Europe’s broader pharmaceutical regulatory environment.
Understanding how these authorities evolved and how they function today helps pharmaceutical companies:
- Build stronger market entry strategies
- Prepare more organized submissions
- Anticipate country-specific expectations
- Improve lifecycle management coordination
- Reduce avoidable submission delays
As pharmaceutical regulation becomes increasingly interconnected across global markets, companies must balance regional harmonization with country-specific compliance requirements.
Without a structured regulatory strategy, differences between agencies can create administrative inefficiencies, inconsistent documentation, and increased operational complexity.
Common Challenges in Multi-Agency Regulatory Management
Managing submissions across multiple European authorities often requires companies to coordinate:
- Different regulatory timelines
- Country-specific administrative documentation
- Product information updates
- Pharmacovigilance obligations
- Lifecycle management activities
- Submission formatting expectations
As companies expand into additional European and international markets, maintaining consistency across regulatory records becomes increasingly important. Even small differences between submissions, timelines, or administrative requirements can create avoidable delays and additional regulatory pressure.
Well-organized submission processes help companies maintain stronger visibility across regulatory activities while reducing the risk of inconsistencies between authorities.
For many pharmaceutical companies, managing these responsibilities internally can become resource intensive, particularly when submissions involve multiple markets, evolving regulatory expectations, and ongoing lifecycle activities.
Navigating Europe’s Evolving Regulatory Landscape
A seasoned global regulatory expert, Registrar Corp helps pharmaceutical companies navigate complex European regulatory activities with greater clarity and confidence through our European regulatory support services.
Our team supports companies through:
- Multi-country submission management
- eCTD publishing support
- Administrative updates and lifecycle activities
- Regulatory document organization
- ASMF & CEP management support
Our regulatory experts help simplify submission preparation by improving document consistency, supporting more efficient regulatory coordination, and helping companies maintain stronger control over regulatory documentation and procedural workflows.
We also provide secure access to RegistrarHub, our online document repository designed to help companies maintain centralized access to important regulatory documentation and submission records.
As European and international regulatory frameworks continue to evolve, companies that invest in structured regulatory processes and experienced submission support are better positioned to maintain compliance, reduce administrative complexity, and support sustainable market growth across multiple jurisdictions.
